
杜洋教授团队在Nature和Science子刊分别发表最新研究成果
原标题:杜洋教授团队在Nature和Science子刊分别发表人体生物钟调控和肾上腺素靶点的最新研究成果
近日,香港中文大学(深圳)生命与健康科学学院、医学院、科比尔卡创新药物开发研究院杜洋教授团队在国际一流科技期刊Nature Communication和Science Advances杂志上分别发表了最新研究成果。其中,杜洋教授团队在Nature Communications杂志揭示人体生物钟调控褪黑素受体的信号传导机制,在Science子刊Science Advances揭示了人体肾上腺素受体药物作用的分子机制,其科研成果将为相关疾病的治疗与药物开发带来曙光。
以下为杜洋教授团队在Nature和Science子刊分别发表的两个具体研究成果:
杜洋教授团队在Science子刊Science Advances揭示人体肾上腺素受体药物作用的分子机制
香港中文大学(深圳)生命与健康科学学院、医学院、科比尔卡创新药物开发研究院杜洋教授,柳正教授联合德国埃朗根-纽伦堡大学国际著名药理学家Peter Gmeiner教授研究团队以及前清华大学医学院袁道鹏博士,在国际一流科技期刊Science Advances杂志(IF=14.136)发表了最新的研究成果:“Structural insights into ligand recognition, activation, and signaling of the α2A adrenergic receptor”。香港中文大学(深圳)杜洋教授、柳正教授及纽伦堡大学Peter Gmeiner教授为本文共同通讯作者。香港中文大学(深圳)生命与健康科学学院、科比尔卡创新药物开发研究院研究助理徐俊(现美国斯坦福大学Brian Kobilka教授实验室博士后)和研究助理曹昇为论文共同第一作者,香港中文大学(深圳)为第一单位。该研究采用冷冻电镜技术首次解析了α2A-GoA复合体分别结合咪唑啉类(imidazoline derivatives)激动剂(溴莫尼定Brimonidine、右美托咪啶Dexmedetomidine和羟间唑啉Oxymetazoline),以及结合内源性荷尔蒙去甲肾上腺素(norepinephrine)的三维结构。

人体超级能量激素-肾上腺素生理功能
肾上腺素能受体(Adrenergic receptor)是一种受内源性儿茶酚胺(肾上腺素,去甲肾上腺素)激活,介导生理活动的G蛋白偶联受体(G-protein coupled receptor, GPCR)【1,2】。在哺乳动物中,肾上腺素能受体共有9个成员,分为三类:α1(α1A, α1B和α1D)类受体,α2(α2A, α2B和α2C)类受体,和β(β1,β2,β3)类受体【2,3】,其中α2类受体能够调节一系列生理功能,包括心率、血压、血糖调节、胰岛素稳态和镇痛作用,同时介导去甲肾上腺素能神经末端突触前膜神经递质释放的反馈抑制【4-6】。α2类受体主要与Gi/o偶联抑制环磷酸腺苷(CAMP)的产生【4,7】,也有证据表明其能和Gs偶联【7-10】,同时该类受体也可通过激动剂相关的GRK(G 蛋白偶联受体激酶)磷酸化作用和β抑制蛋白偶联,实现受体内化和独立于G蛋白的信号传导【11-13】。过去的几十年里,科学家开发了很多α2类受体的激动剂,其中部分被用于麻醉、痛觉管理和高血压治疗【14,15】。但是相比于药物作用的分子基础被充分研究的β类受体【16-21】,α2类受体药物作用和激活机理的结构基础尚不明确。α2类受体和不同激动剂结合的结构研究将为G蛋白偶联的特异性提供新视角,为新激动剂开发提供理论依据。
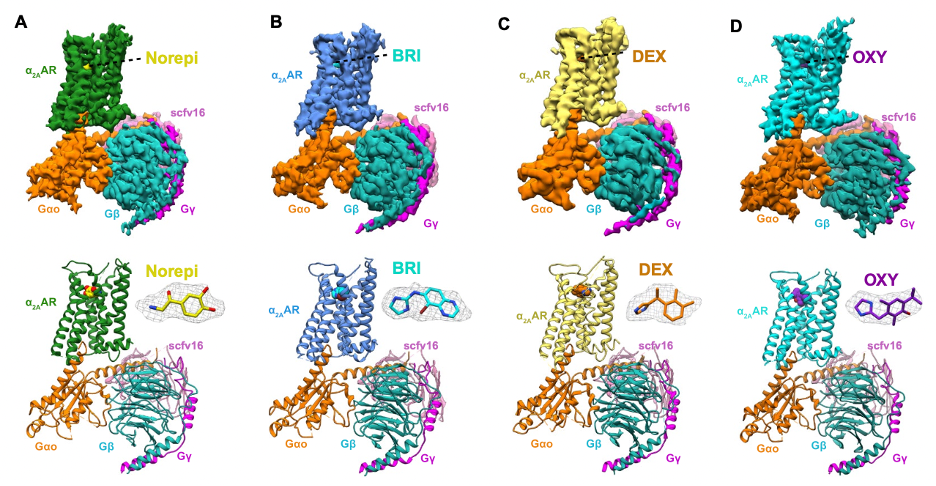
alpha2AR肾上腺素受体与不同药物结合的高分辨率结构
本研究对肾上腺素能受体的配体识别、激活机理以及信号传导选择等都具有广泛的影响,列举如下:(1)揭示了α2AAR的正位结合口袋(orthosteric binding pocket,OBP)识别内源性荷尔蒙去甲肾上腺素及咪唑啉类激动剂的机制;(2)揭示了内源性荷尔蒙去甲肾上腺素对于α2类受体亲和力高于β类受体的机理;(3)发现了在受体激活过程中重要的芳香性开关;(4)发现了和β类受体不同的、与激活相关的结构变化,但共有正构口袋和G蛋白偶联区的变构机理;(5)提出了α2AAR对G蛋白、β抑制蛋白偶联和选择性的关键区域和残基。值得一提的是,研究中使用的咪唑啉类激动剂,溴莫尼定、右美托咪啶和羟间唑啉均为上市药物,分别用于治疗开角型青光眼及高眼压、镇静止痛和减轻炎症所致的充血和水肿。其中羟间唑啉在对α2AAR残基Y4096.55的突变实验中,有别于其他两个激动剂及去甲肾上腺素对于G蛋白、β抑制蛋白招募效能的降低,尤其展现出了更高的β抑制蛋白招募效能,表明该残基在偏向信号传导(biased signaling)中扮演复杂和重要的角色。无独有偶,最近对于β2AR 6.55位置的结构功能研究和计算研究也表明该位置的残基在调控偏向中的重要地位【20,22】。该发现为偏向药物(biased drug)的分子机理研究和药物开发提供了新的切入点。
综上所述,该团队利用单颗粒冷冻电镜技术解析了肾上腺素能受体α2AAR与G蛋白的复合物结构,从而在原子层面上详细阐释了α2AAR的配体识别、别构调节及与G蛋白偶联的机制,该项研究将促进基于α2AAR结构的药物研究,为镇痛药物开发和高血压治疗带来新的曙光。
杜洋教授团队在Nature Communications揭示人体生物钟调控褪黑素受体的信号传导机制
香港中文大学(深圳)生命与健康科学学院、医学院、科比尔卡创新药物开发研究院杜洋教授,柳正教授联合中国科学技术大学陶余勇教授研究团队,在国际一流科技期刊Nature Communication杂志(IF=14.919)发表了最新的研究成果:“Structural basis of the ligand binding and signaling mechanism of melatonin receptors”。香港中文大学(深圳)杜洋教授、柳正教授及中国科学技术大学陶余勇教授为本文共同通讯作者。香港中文大学(深圳)生命与健康科学学院、科比尔卡创新药物开发研究院访问博士生王庆功,香港中文大学(深圳)生命与健康科学学院、科比尔卡创新药物开发研究院技术员卢秋远为论文共同第一作者。该研究采用冷冻电镜技术解析了MT1-Gi复合体分别结合激动剂2-碘褪黑激素(2-iodomelatonin)、雷美替胺(ramelteon),以及MT2-Gi复合体结合ramelteon三维结构,并阐明了褪黑素和药物的激活机制。
人体生物钟调节激素-褪黑素调控昼夜节律
在脊椎动物中,褪黑素激素主要由松果体分泌,遵循昼夜节律的分泌模式。通过作用于褪黑激素受体 (MT),在调节机体昼夜节律和维持稳定睡眠模式中发挥重要的作用(1, 2)。此外,越来越多研究表明褪黑素-MT信号通路的激活还可以调节其他生理过程,包括调节心血管系统和免疫系统(3, 4)。该信号通路激活在抑制癌症、促进骨形成、调节糖代谢以及调剂神经退行性疾病中的作用也得到了证实(5-7)。褪黑素受体是一类既可以在中枢神经系统 (CNS),又外周组织中表达的class A GPCR的亚家族。在人类中,MT 家族由两个结构序列高度保守的成员组成,包括MT1 和 MT2受体,它们在整个序列和跨膜部分序列分别具有 55% 和 70%相似性。MT1 和 MT2 都主要通过与Gi/o 蛋白偶联,从而抑制腺苷酸环化酶活性并降低细胞内 3',5'-环磷酸腺苷 (adenosine 3’,5’-cyclic monophosphate;cAMP) 的浓度,去发挥其重要的生理作用。尽管目前有报道MT1受体还可以结合下游Gq/11蛋白,但是目前关于与G 蛋白家族确切的偶联谱仍不清楚。在人大脑中,MT1 主要在蓝斑和下丘脑外侧等重要神经功能区中表达于REM 区域)(8, 9),参与调节睡眠中警觉状态的快速眼动 (REM) 阶段(10);但 MT2 主要在网状丘脑合成及分泌(NREM 区域)(8, 9),可以选择性地调节非快速眼动 (NREM) 睡眠阶段(10, 11),此外MT2还与 2 型糖尿病 (type-2 diabetes;T2D)形成相关。鉴于 MT1 和 MT2 的不同生理作用,获得选择性配体具有重要应用前景。然而,市场上或临床评估中的大多数药物,如雷美替昂, 他司美替昂和阿戈美拉汀,都是非选择性的(12, 13)。因此,通过解析 MTs-配体结合口袋结构信息,对开发高效的选择性激动剂药物提供理论依据,具有重要研究价值。
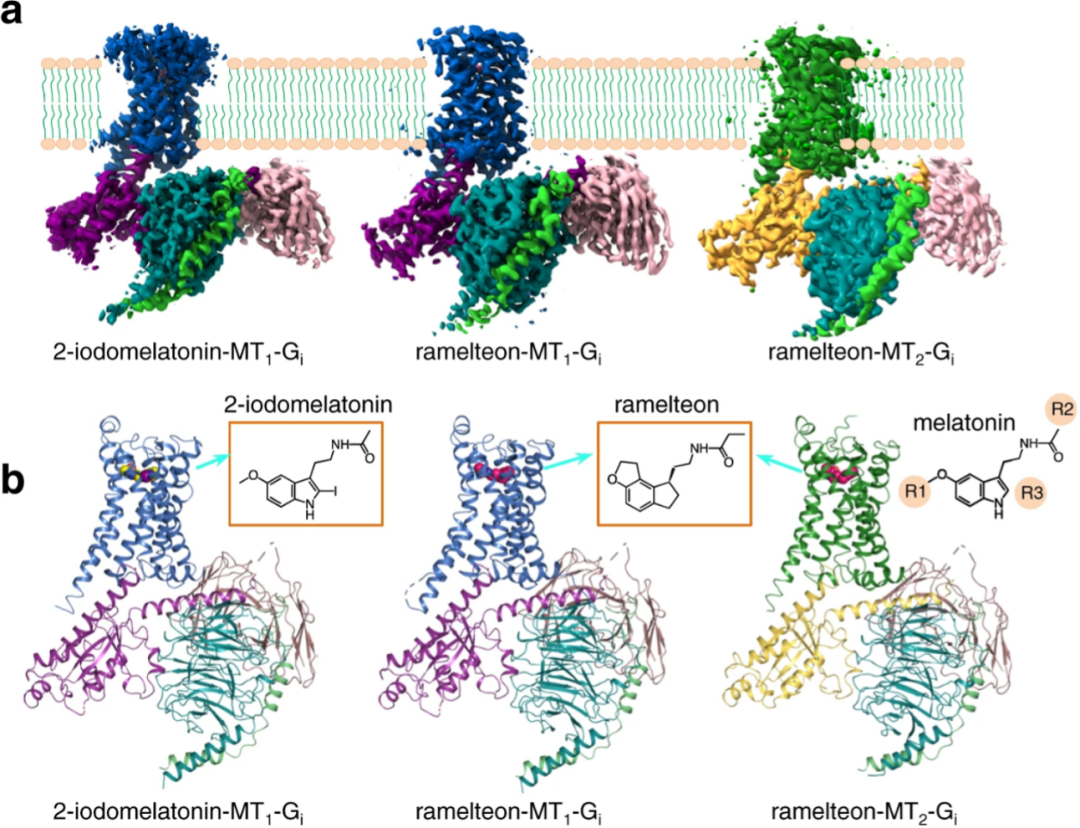
褪黑素受体与不同药物结合的高分辨率结构
本研究对褪黑素受体的配体识别、激活机理以及信号转导选择等都具有广泛的影响,列举如下:(1)发现了H5.46, N4.60, 及 Y5.38三个残基位点在MT受体正位结合口袋(orthosteric binding pocket,OBP)识别激动剂配体中的起到重要作用;(2)揭示了MT1与MT2的OBP与激动剂结合状态下结构差异及特征,通过信号转导药理学实验和分子动力学模拟对该位点进行了验证,为设计有效靶向小分子药物的设计提供了理论基础,具有重要意义;(3)通过与inactive状态下比较,阐释褪黑素受体激活机制,通过生理生化实验进一步验证关键的残基位点。(4)解析了MT1与MT2在激动剂激活状态下结构上的差异, 提出了MT受体对Gi蛋白偶联和选择性的关键残基,进一步分析与下游G蛋白结合位点,发现第二个胞内环(ICL2)构象具有明显差异,这初步解释MT1受体可以结合下游Gq/11蛋白,而MT2受体不能结合的原因。为GPCR选择性激活下游不同G蛋白的机制提供理论基础。
综上所述,该团队利用单颗粒冷冻电镜技术解析了褪黑素受体MT1及MT2与G蛋白的复合物结构,从而在原子层面上详细阐释了褪黑素受体的配体识别、别构调节及与G蛋白偶联的机制,该项研究将为失眠症、情绪障碍和癌症等疾病的高选择性药物开发和治疗带来新的曙光。
教授简介

杜洋
生命与健康科学学院、医学院助理教授
教育背景
博士(中国科学技术大学)
学士 (中南大学)
研究领域
G蛋白偶联受体、受体生物学、信号转导、结构药理学、基于结构的药物设计
杜洋教授现为香港中文大学(深圳)生命与健康科学学院、医学院助理教授,博士生导师,校长学者,教育部青年,广东省珠江青年拔尖人才。他还是香港中文大学(深圳)科比尔卡创新药物开发研究院责任研究员,领导团队从事GPCR的转化研究和新药开发工作。
杜洋教授在2011年于中国科学技术大学取得博士学位。之后赴美国斯坦福大学医学院,师从2012年诺贝尔化学奖得主Brian Kobilka教授从事G蛋白偶联受体(GPCR)相关的博士后研究工作。2016年提升为斯坦福医学中心固定职位的研究科学家。期间获美国心脏协会全额博士后奖学金、斯坦福心血管研究所资助和密歇根大学安娜堡医学院药学系tenure-track助理教授职位等。
主要研究方向是以重要药物靶点GPCR为对象,从事其与下游信号分子复合物的结构、功能和药物发现工作。迄今已发表50多篇高质量SCI论文,包括以第一或通讯作者(含共同)在Cell、Sci. Adv.、JACS、Nature Comm.、Cell Res.等国际一流期刊报道的一系列科研成果,作为无成瘾止痛药开发的美国专利共同发明人。并受邀为Science、Nature Comm.等多种国际顶尖科学杂志审稿,同时是国自然交叉科学部和教育部项目评阅人。



